solo para uso en investigación
PRN1371 FGFR inhibidor
Cat. No.S8578
Estructura química
Peso molecular: 561.46
Control de calidad
| Dianas relacionadas | EGFR VEGFR JAK PDGFR Src HIF FLT FLT3 HER2 Bcr-Abl |
|---|---|
| Otros FGFR Inhibidores | PD173074 AZD4547 (Fexagratinib) BLU9931 Futibatinib (TAS-120) LY2874455 PD-166866 Zoligratinib (Debio-1347) H3B-6527 Fisogatinib (BLU-554) SSR128129E |
Información química, almacenamiento y estabilidad
| Peso molecular | 561.46 | Fórmula | C26H30Cl2N6O4 |
Almacenamiento (Desde la fecha de recepción) | |
|---|---|---|---|---|---|
| Nº CAS | 1802929-43-6 | Descargar SDF | Almacenamiento de soluciones madre |
|
|
| Sinónimos | N/A | Smiles | CNC1=NC=C2C=C(C(=O)N(C2=N1)CCCN3CCN(CC3)C(=O)C=C)C4=C(C(=CC(=C4Cl)OC)OC)Cl | ||
Solubilidad
|
In vitro |
DMSO
: 56 mg/mL
(99.73 mM)
Water : Insoluble Ethanol : Insoluble |
Calculadora de Molaridad
|
In vivo |
|||||
Calculadora de formulación in vivo (Solución clara)
Paso 1: Introduzca la información a continuación (Recomendado: Un animal adicional para tener en cuenta la pérdida durante el experimento)
Paso 2: Introduzca la formulación in vivo (Esto es solo la calculadora, no la formulación. Por favor, contáctenos primero si no hay una formulación in vivo en la sección de Solubilidad.)
Resultados del cálculo:
Concentración de trabajo: mg/ml;
Método para preparar el líquido maestro de DMSO: mg fármaco predissuelto en μL DMSO ( Concentración del líquido maestro mg/mL, Por favor, contáctenos primero si la concentración excede la solubilidad del DMSO del lote del fármaco. )
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadirμL PEG300, mezclar y clarificar, luego añadirμL Tween 80, mezclar y clarificar, luego añadir μL ddH2O, mezclar y clarificar.
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadir μL Aceite de maíz, mezclar y clarificar.
Nota: 1. Por favor, asegúrese de que el líquido esté claro antes de añadir el siguiente disolvente.
2. Asegúrese de añadir el (los) disolvente(s) en orden. Debe asegurarse de que la solución obtenida, en la adición anterior, sea una solución clara antes de proceder a añadir el siguiente disolvente. Se pueden utilizar métodos físicos como el vórtice, el ultrasonido o el baño de agua caliente para ayudar a la disolución.
Mecanismo de acción
| Targets/IC50/Ki |
FGFR1
(Cell-free assay) 0.6 nM
FGFR2
(Cell-free assay) 1.3 nM
FGFR3
(Cell-free assay) 4.1 nM
CSF1R
(Cell-free assay) 8.1 nM
FGFR4
(Cell-free assay) 19.3 nM
|
|---|---|
| In vitro |
PRN1371 es un inhibidor nanomolar irreversible de FGFR1-4. Este compuesto presenta un perfil único de alta potencia bioquímica y celular (FGFR1 IC50 = 0,6 nM, SNU16 IC50 = 2,6 nM), un compromiso prolongado con el objetivo (ocupación de FGFR1 24 h = 96 %), una inhibición de hERG <30 % a 1 μM, y una buena estabilidad ADME predicha con reactividad BME Kd>100 μM. Este químico mantuvo una alta ocupación de FGFR1 con solubilidad mejorada y excepcional biodisponibilidad oral.
|
| In vivo |
Un estudio PK intravenoso (2 mg/kg) de PRN1371 en ratas mostró un aclaramiento rápido (Cl = 160 ml/min/kg), sin embargo, la administración oral (20 mg/kg) demostró una alta exposición oral (AUC = 4348 h·ng/mL) y una vida media razonable (t1/2 = 3,8 h). Los estudios de PK de este compuesto en ratas, perros y monos cinomolgos mostraron un aclaramiento intravenoso rápido en todas las especies; sin embargo, hubo grandes diferencias entre especies en la exposición oral y la biodisponibilidad para el mono en comparación con la rata y el perro. En la rata, la alta exposición tras la dosificación oral (p. ej., Cmax = 1785 ng/mL, AUC = 4348 ng·h/mL) y una biodisponibilidad (F) >100 % sugirieron una buena absorción y saturación parcial de los mecanismos de aclaramiento con la dosis de 20 mg/kg. Exclusivamente en la rata, existe una gran diferencia en la vida media entre las vías de administración intravenosa (t1/2 = 0,8 h) y oral (t1/2 = 3,8 h), lo que también indica una posible saturación de un mecanismo de aclaramiento tras la administración oral. En los perros, la misma formulación de suspensión de metilcelulosa utilizada para la rata dio una baja absorción oral y biodisponibilidad (F < 15 %). En el modelo de ratón de xenoinjerto de cáncer gástrico SNU16, este compuesto indujo una reducción dosis-dependiente del volumen tumoral y hasta un 68 % de inhibición del crecimiento tumoral a la dosis más alta de 10 mg/kg b.i.d. después de 27 días de tratamiento. Todas las dosis fueron bien toleradas sin pérdida significativa de peso corporal.
|
Referencias |
Aplicaciones
| Métodos | Biomarcadores | Imágenes | PMID |
|---|---|---|---|
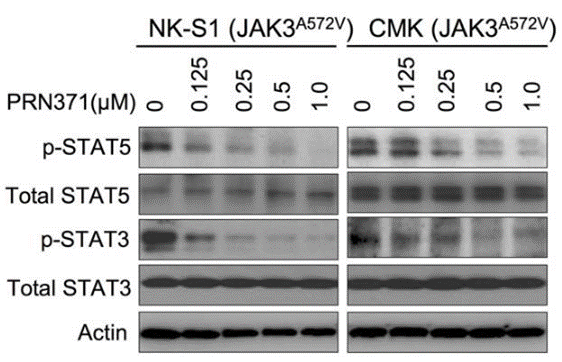
| Western blot | p-STAT5 / STAT5 / p-STAT3 / STAT3 |
|
29434279 |
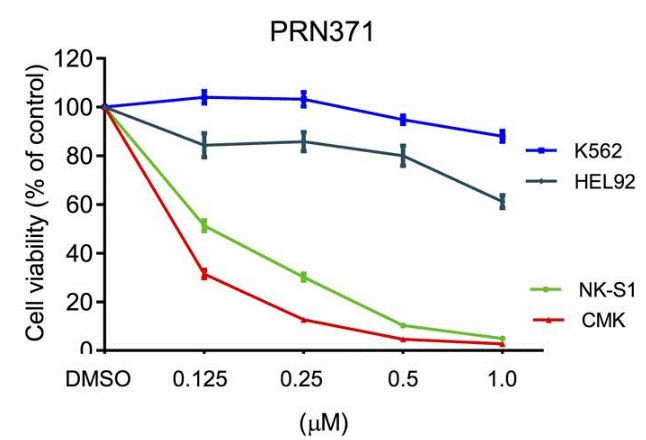
| Growth inhibition assay | Cell viability |
|
29434279 |
Información del ensayo clínico
(datos de https://clinicaltrials.gov, actualizado el 2024-05-22)
| Número NCT | Reclutamiento | Condiciones | Patrocinador/Colaboradores | Fecha de inicio | Fases |
|---|---|---|---|---|---|
| NCT02608125 | Terminated | Solid Tumors|Metastatic Urothelial Carcinoma & Renal Pelvis & Ureter |
Principia Biopharma a Sanofi Company |
October 28 2015 | Phase 1 |
Soporte técnico
Tel: +1-832-582-8158 Ext:3
Si tiene alguna otra consulta, por favor deje un mensaje.
Los productos son solo para uso de investigación. No para uso humano. No vendemos a pacientes.
©Copyright 2013 Selleck Chemicals. Todos los derechos reservados.