solo para uso en investigación
MK-8617 HIF modulador
Cat. No.S8443
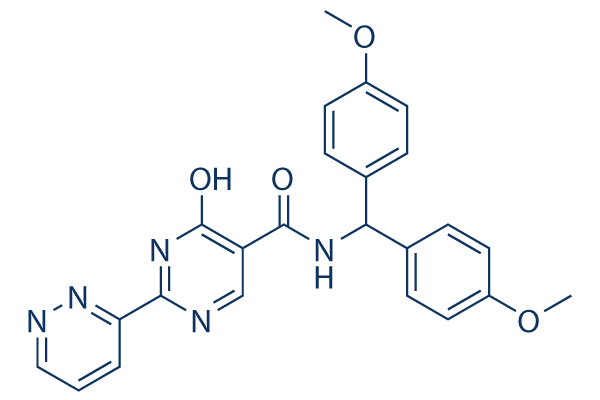
Estructura química
Peso molecular: 443.45
Control de calidad
| Dianas relacionadas | EGFR VEGFR JAK PDGFR FGFR Src FLT FLT3 HER2 Bcr-Abl |
|---|---|
| Otros HIF Inhibidores | PT2399 PX-478 Dihydrochloride BAY 87-2243 KC7F2 Lificiguat (YC-1) IOX2 CAY10585 (LW 6) Molidustat (BAY 85-3934) PT2385 IDF-11774 |
Información química, almacenamiento y estabilidad
| Peso molecular | 443.45 | Fórmula | C24H21N5O4 |
Almacenamiento (Desde la fecha de recepción) | |
|---|---|---|---|---|---|
| Nº CAS | 1187990-87-9 | Descargar SDF | Almacenamiento de soluciones madre |
|
|
| Sinónimos | N/A | Smiles | COC1=CC=C(C=C1)C(C2=CC=C(C=C2)OC)NC(=O)C3=CN=C(NC3=O)C4=NN=CC=C4 | ||
Solubilidad
|
In vitro |
DMSO
: 10 mg/mL
(22.55 mM)
Water : Insoluble Ethanol : Insoluble |
Calculadora de Molaridad
|
In vivo |
|||||
Calculadora de formulación in vivo (Solución clara)
Paso 1: Introduzca la información a continuación (Recomendado: Un animal adicional para tener en cuenta la pérdida durante el experimento)
Paso 2: Introduzca la formulación in vivo (Esto es solo la calculadora, no la formulación. Por favor, contáctenos primero si no hay una formulación in vivo en la sección de Solubilidad.)
Resultados del cálculo:
Concentración de trabajo: mg/ml;
Método para preparar el líquido maestro de DMSO: mg fármaco predissuelto en μL DMSO ( Concentración del líquido maestro mg/mL, Por favor, contáctenos primero si la concentración excede la solubilidad del DMSO del lote del fármaco. )
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadirμL PEG300, mezclar y clarificar, luego añadirμL Tween 80, mezclar y clarificar, luego añadir μL ddH2O, mezclar y clarificar.
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadir μL Aceite de maíz, mezclar y clarificar.
Nota: 1. Por favor, asegúrese de que el líquido esté claro antes de añadir el siguiente disolvente.
2. Asegúrese de añadir el (los) disolvente(s) en orden. Debe asegurarse de que la solución obtenida, en la adición anterior, sea una solución clara antes de proceder a añadir el siguiente disolvente. Se pueden utilizar métodos físicos como el vórtice, el ultrasonido o el baño de agua caliente para ayudar a la disolución.
Mecanismo de acción
| Targets/IC50/Ki |
PHD1
(Cell-free assay) 1 nM
PHD2
(Cell-free assay) 1 nM
PHD3
(Cell-free assay) 14 nM
|
|---|---|
| In vitro |
MK-8617 no es un inhibidor significativo de las enzimas del citocromo p450 in vitro (IC50), CYP1A2, 3A4, 2B6, 2C9, 2C19 o 2D6, >60 μM, y es un inhibidor reversible moderado de CYP2C8 a 1,6 μM in vitro. Este compuesto es inactivo cuando se evalúa a 10 μM contra un panel general de 171 ensayos de unión a radioligandos y enzimáticos. |
| In vivo |
El MK-8617 tritiado exhibe un recambio metabólico mínimo en microsomas hepáticos (+NADPH) de rata, perro y mono (<10% de recambio) pero un recambio significativo en microsomas hepáticos humanos (34% de recambio) después de 60 min (compuesto de 10 μM, 1 mg/mL de proteína microsomal). En cuanto a su perfil farmacocinético, este compuesto muestra una buena biodisponibilidad oral en todas las especies (36-71%), con un bajo aclaramiento y volumen de distribución. El compuesto aún tiene una vida media de eliminación relativamente larga en especies preclínicas. En ratones (C57Bl/6), dosis únicas de 5 y 15 mpk po (n = 3) causan aumentos en los reticulocitos circulantes medidos tanto a los 3 como a los 4 días después del desafío con el compuesto. En ratas (Sprague-Dawley), una titulación de dosis única de 1,5, 5 y 15 mpk po (n = 5) causa un gran aumento en los niveles séricos de eritropoyetina (EPO) de 1,7-, 8- y 204-veces en relación con el vehículo, respectivamente. Se observan aumentos en los reticulocitos circulantes a 5 y 15 mg/kg 3 días después del desafío y, con la dosis de 15 mg, 4 días después del desafío. |
Referencias |
|
Soporte técnico
Tel: +1-832-582-8158 Ext:3
Si tiene alguna otra consulta, por favor deje un mensaje.
Los productos son solo para uso de investigación. No para uso humano. No vendemos a pacientes.
©Copyright 2013 Selleck Chemicals. Todos los derechos reservados.