solo para uso en investigación
Fenebrutinib (GDC-0853) BTK inhibidor
Cat. No.S8421
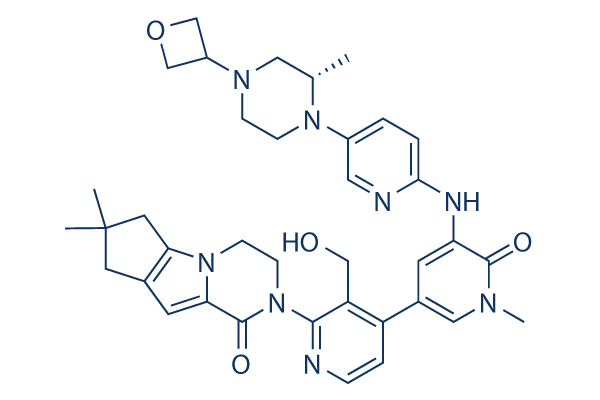
Estructura química
Peso molecular: 664.80
Control de calidad
- Citado en Nature Medicine por su máxima calidad
- COA
- NMR
- SDS
- Hoja de datos
| Dianas relacionadas | EGFR VEGFR PDGFR FGFR c-Met Src MEK CSF-1R FLT3 HER2 |
|---|---|
| Otros BTK Inhibidores | Catadegbrutinib (BGB-16673) Spebrutinib (AVL-292) tirabrutinib(ONO-4059) hydrochloride CGI1746 LFM-A13 CNX-774 Evobrutinib BMS-935177 Branebrutinib (BMS-986195) Pirtobrutinib (LOXO-305) |
Información química, almacenamiento y estabilidad
| Peso molecular | 664.80 | Fórmula | C37H44N8O4 |
Almacenamiento (Desde la fecha de recepción) | 3 years -20°C powder |
|---|---|---|---|---|---|
| Nº CAS | 1434048-34-6 | -- | Almacenamiento de soluciones madre |
|
|
| Sinónimos | N/A | Smiles | CC1CN(CCN1C2=CN=C(C=C2)NC3=CC(=CN(C3=O)C)C4=C(C(=NC=C4)N5CCN6C7=C(CC(C7)(C)C)C=C6C5=O)CO)C8COC8 | ||
Solubilidad
|
In vitro |
DMSO
: 8 mg/mL
(12.03 mM)
Water : Insoluble Ethanol : Insoluble |
Calculadora de Molaridad
|
In vivo |
|||||
Calculadora de formulación in vivo (Solución clara)
Paso 1: Introduzca la información a continuación (Recomendado: Un animal adicional para tener en cuenta la pérdida durante el experimento)
Paso 2: Introduzca la formulación in vivo (Esto es solo la calculadora, no la formulación. Por favor, contáctenos primero si no hay una formulación in vivo en la sección de Solubilidad.)
Resultados del cálculo:
Concentración de trabajo: mg/ml;
Método para preparar el líquido maestro de DMSO: mg fármaco predissuelto en μL DMSO ( Concentración del líquido maestro mg/mL, Por favor, contáctenos primero si la concentración excede la solubilidad del DMSO del lote del fármaco. )
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadirμL PEG300, mezclar y clarificar, luego añadirμL Tween 80, mezclar y clarificar, luego añadir μL ddH2O, mezclar y clarificar.
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadir μL Aceite de maíz, mezclar y clarificar.
Nota: 1. Por favor, asegúrese de que el líquido esté claro antes de añadir el siguiente disolvente.
2. Asegúrese de añadir el (los) disolvente(s) en orden. Debe asegurarse de que la solución obtenida, en la adición anterior, sea una solución clara antes de proceder a añadir el siguiente disolvente. Se pueden utilizar métodos físicos como el vórtice, el ultrasonido o el baño de agua caliente para ayudar a la disolución.
Mecanismo de acción
| Targets/IC50/Ki |
BTK
(Cell-free assay) 0.91 nM(Ki)
BTK C481R
(Cell-free assay) 1.3 nM(Ki)
BTK C481S
(Cell-free assay) 1.6 nM(Ki)
BTK T474M
(Cell-free assay) 3.4 nM(Ki)
BTK T474I
(Cell-free assay) 12.6 nM(Ki)
|
|---|---|
| In vitro |
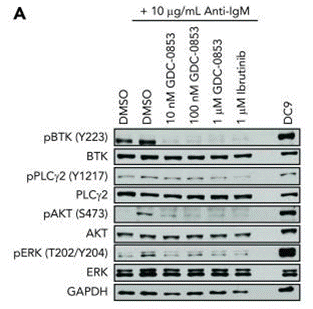
Cuando se probó a 1 μM contra un amplio panel de ensayos bioquímicos de quinasas humanas, Fenebrutinib (GDC-0853) inhibe solo 3 de 286 quinasas fuera del objetivo. Basado en los valores de IC50 determinados, la selectividad para Btk es >100 veces mayor contra cada una de estas 3 dianas secundarias: Bmx (153 veces), Fgr (168 veces) y Src (131 veces). Este compuesto bloquea tanto la señalización del BCR de las células B como la señalización del FcγR de los monocitos. En el ensayo enzimático bioquímico de Btk in vitro, muestra un tiempo de residencia promedio con Btk de 18,3 ± 2,8 horas. Bloquea la autofosforilación celular de la Btk de tipo salvaje (WT Btk) y del mutante C481S. Las células de LLC (leucemia linfocítica crónica) tratadas con GDC-0853 in vitro antes de la estimulación del BCR demuestran niveles reducidos de fosforilación de BTK y una activación disminuida de los objetivos posteriores, incluyendo PLCγ2, AKT y ERK. Inhibe la transcripción dependiente de NF-κB, reduce la activación y afecta la migración. Este compuesto carece de inhibición de EGFR e ITK en el sistema celular y no afecta la activación del receptor de células T. |
| Ensayo de quinasa |
Selectividad de la quinasa
|
|
La selectividad de la quinasa Fenebrutinib (GDC-0853) se evalúa a una concentración de 1 µM en un panel de hasta 287 ensayos de actividad y unión de quinasas humanas recombinantes, incluyendo tirosina quinasas citoplasmáticas y de receptores, serina/treonina quinasas y quinasas lipídicas. Los ensayos de actividad de la quinasa miden la fosforilación de péptidos o la producción de ADP, mientras que los ensayos de unión monitorizan el desplazamiento de las sondas de unión al sitio de ATP. Las concentraciones de ATP utilizadas en los ensayos de actividad suelen estar dentro de 2 veces el valor de la constante de Michaelis aparente (Kmapp) determinada experimentalmente para cada quinasa, mientras que las concentraciones de trazador de unión competitiva utilizadas en los ensayos de unión suelen estar dentro de 3 veces los valores de la constante de disociación (Kd) determinada experimentalmente. Este compuesto se prueba por duplicado contra cada quinasa y se informan los valores medios de % de inhibición. Para las quinasas que se inhiben en cerca o más del 80% a la concentración de prueba, se realizan titulaciones de inhibidores de 10 puntos utilizando los mismos ensayos para determinar las concentraciones de inhibidores que causaron el 50% de inhibición (IC50).
|
|
| In vivo |
Fenebrutinib (GDC-0853) tiene un aclaramiento moderado de 27,4 mL/min/kg y una excelente biodisponibilidad (F=65%) en ratas a las que se administró 0,2 mg/kg mediante inyección intraperitoneal o 1 mg/kg PO. Su aclaramiento plasmático es de 27,4 mL/min/kg, el volumen de distribución (Vd) es de 5,42 L/kg y la semivida plasmática (t1/2) es de 2,2 h. Este compuesto también demuestra propiedades PK favorables en perros. La semivida de 3,8 horas (Clp 10,9 mL/min/kg, Vd 2,96 L/kg) y la alta biodisponibilidad oral (85%) también permiten alcanzar exposiciones suficientes en estudios de toxicología en perros. Es bien tolerado tanto en ratas como en perros y presenta un perfil de seguridad globalmente favorable. GDC-0853 es útil en el tratamiento de la artritis reumatoide y otras enfermedades autoinmunes mediadas por células B o mieloides. En un estudio de dosis única ascendente (SAD) (0,5 mg a 600 mg) y un estudio de dosis múltiples ascendentes (MAD) durante 14 días (250 mg BID a 500 mg QD), fue muy bien tolerado sin eventos adversos graves, sin señales de seguridad y sin toxicidades limitantes de la dosis. Se absorbió bien y mostró una farmacocinética lineal y dosis-proporcional. En ratas Sprague-Dawley (SD), la administración de este compuesto y otros inhibidores de BTK estructuralmente diversos durante 7 días o más causa lesiones pancreáticas que consisten en hemorragia multifocal centrada en los islotes, inflamación, fibrosis y macrófagos cargados de pigmento con atrofia, degeneración e inflamación adyacente de las células acinares exocrinas lobulillares. No se observaron hallazgos similares en ratones o perros a exposiciones mucho más altas. |
Referencias |
|
Aplicaciones
| Métodos | Biomarcadores | Imágenes | PMID |
|---|---|---|---|
| Western blot | p-BTK / BTK / p-PLCγ2 / PLCγ2 / p-AKT / AKT / p-ERK / ERK |
|
30018078 |
Información del ensayo clínico
(datos de https://clinicaltrials.gov, actualizado el 2024-05-22)
| Número NCT | Reclutamiento | Condiciones | Patrocinador/Colaboradores | Fecha de inicio | Fases |
|---|---|---|---|---|---|
| NCT05119569 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 1 2022 | Phase 2 |
| NCT04586023 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 24 2021 | Phase 3 |
| NCT04586010 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 17 2021 | Phase 3 |
| NCT03693625 | Terminated | Urticaria |
Genentech Inc. |
September 27 2018 | Phase 2 |
| NCT03596632 | Completed | Healthy Participants |
Hoffmann-La Roche |
July 27 2018 | Phase 1 |
Soporte técnico
Tel: +1-832-582-8158 Ext:3
Si tiene alguna otra consulta, por favor deje un mensaje.
Los productos son solo para uso de investigación. No para uso humano. No vendemos a pacientes.
©Copyright 2013 Selleck Chemicals. Todos los derechos reservados.