solo para uso en investigación
Ticagrelor P2 Receptor antagonista
Cat. No.S4079
Estructura química
Peso molecular: 522.57
Control de calidad
| Dianas relacionadas | Adrenergic Receptor AChR 5-HT Receptor COX Calcium Channel Histamine Receptor Dopamine Receptor GABA Receptor TRP Channel Cholinesterase (ChE) |
|---|---|
| Otros P2 Receptor Inhibidores | A-438079 Hydrochloride A-804598 MRS 2578 A-740003 5-BDBD Gefapixant AF-353 A-317491 Minodronic acid Diquafosol Tetrasodium |
Cultivo celular, tratamiento y concentración de trabajo
| Líneas celulares | Tipo de ensayo | Concentración | Tiempo de incubación | Formulación | Descripción de la actividad | PMID |
|---|---|---|---|---|---|---|
| H9c2 | Function assay | 0.1, 0,3 and 1 μM | 24 h | effective in reducing NCX1 reverse activity when lower concentrations | ||
| EAhy926 | Apoptosis assay | 40 μM and 60 μM | decrease ox-LDL-induced apoptosis, particularly at a higher concentration | |||
| AsPC-1 | Function assay | 10 μM | 2 h | ticagrelor negated the platelet releasate effect on Akt, Erk activation and Slug upregulation | ||
| BxPC-3 | Function assay | 10 μM | 2 h | ticagrelor negated the platelet releasate effect on Akt, Erk activation and Slug upregulation | ||
| Haga clic para ver más datos experimentales de líneas celulares | ||||||
Información química, almacenamiento y estabilidad
| Peso molecular | 522.57 | Fórmula | C23H28F2N6O4S |
Almacenamiento (Desde la fecha de recepción) | |
|---|---|---|---|---|---|
| Nº CAS | 274693-27-5 | Descargar SDF | Almacenamiento de soluciones madre |
|
|
| Sinónimos | AZD6140, AR-C 126532XX | Smiles | CCCSC1=NC(=C2C(=N1)N(N=N2)C3CC(C(C3O)O)OCCO)NC4CC4C5=CC(=C(C=C5)F)F | ||
Solubilidad
|
In vitro |
DMSO
: 100 mg/mL
(191.36 mM)
Water : Insoluble Ethanol : Insoluble |
Calculadora de Molaridad
|
In vivo |
|||||
Calculadora de formulación in vivo (Solución clara)
Paso 1: Introduzca la información a continuación (Recomendado: Un animal adicional para tener en cuenta la pérdida durante el experimento)
Paso 2: Introduzca la formulación in vivo (Esto es solo la calculadora, no la formulación. Por favor, contáctenos primero si no hay una formulación in vivo en la sección de Solubilidad.)
Resultados del cálculo:
Concentración de trabajo: mg/ml;
Método para preparar el líquido maestro de DMSO: mg fármaco predissuelto en μL DMSO ( Concentración del líquido maestro mg/mL, Por favor, contáctenos primero si la concentración excede la solubilidad del DMSO del lote del fármaco. )
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadirμL PEG300, mezclar y clarificar, luego añadirμL Tween 80, mezclar y clarificar, luego añadir μL ddH2O, mezclar y clarificar.
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadir μL Aceite de maíz, mezclar y clarificar.
Nota: 1. Por favor, asegúrese de que el líquido esté claro antes de añadir el siguiente disolvente.
2. Asegúrese de añadir el (los) disolvente(s) en orden. Debe asegurarse de que la solución obtenida, en la adición anterior, sea una solución clara antes de proceder a añadir el siguiente disolvente. Se pueden utilizar métodos físicos como el vórtice, el ultrasonido o el baño de agua caliente para ayudar a la disolución.
Mecanismo de acción
| Características |
First-in-class of a new type of P2Y12 antagonist known as cyclopentyl-triazolo-pyrimidines.
|
|---|---|
| Targets/IC50/Ki |
P2Y12
2 nM(Ki)
|
| In vitro |
Ticicagrelor es un fármaco activo que no requiere activación metabólica después de la absorción intestinal. No compite directamente con el ADP en el sitio de unión del ADP, sino que ocupa un sitio de unión adyacente y actúa de forma alostérica, lo que resulta en un cambio conformacional reversible del receptor. Este compuesto se une reversiblemente al receptor y exhibe un rápido inicio y finalización del efecto. Los estudios de unión en células CHO-K1 transfectadas con el receptor rh-P2Y12 indican que este compuesto exhibe una unión potente, rápida y reversible, con una Kd de 10,5 nM, una kon (constante de asociación) de 0,00011/(nM•s), una koff (constante de disociación) de 0,00087/s, y valores de semivida de 4 min para la unión y 14 min para la desunión, lo que indica que la magnitud de la inhibición plaquetaria depende de las concentraciones del fármaco disponibles para unirse a las plaquetas. Este químico inhibe moderadamente la actividad de CYP2C9 en microsomas hepáticos humanos, mientras que exhibe poca o ninguna inhibición de CYP1A2, CYP2B6, CYP2C8, CYP2C19, CYP2D6 y CYP2E1. En microsomas hepáticos humanos, inhibe la 4-hidroxilación de midazolam, mientras que activa la 1_-hidroxilación de midazolam. Evaluado en hepatocitos humanos frescos, este compuesto no es un inductor de CYP1A2 o CYP3A4. |
| Ensayo de quinasa |
Ensayos de unión utilizando CHO-K1 transfectadas con P2Y12 o membranas de plaquetas humanas
|
|
Se añaden membranas (5 μg de proteína) a una placa de 96 pocillos que contiene [125I]AZ11931285 (125 pM), [3H]ADP (10 nM) o [33P]2MeS-ADP (62,5 pM), la concentración requerida de competidor y un volumen suficiente de tampón (50 mM Tris, 5 mM MgCl2, 50 mM NaCl y 0,1% de BSA libre de nucleótidos, pH 7,4) para llevar el volumen total en cada pocillo a 200 μL. Los estudios de unión utilizando membranas plaquetarias y [3H]ADP se realizan en presencia de 100 μM (concentración final) de MRS2179 para prohibir la unión a P2Y1. Las relaciones señal/ruido para las células CHO-K1 transfectadas con P2Y12 son aproximadamente 14 para [3H]ADP (señal específica: 895 c.p.m.), 24 para [33P]2MeSADP (señal específica: 3308 c.p.m.) y 24 para [125I]AZ11931285 (señal específica: 3308 c.p.m.). Para los estudios que utilizan membranas plaquetarias, las relaciones señal/ruido son aproximadamente 2 para [33P]2MeS-ADP y [3H]ADP y 1,5 para [125I]AZ11931285, con una señal específica entre 100 y 400 c.p.m. En este estudio, se utiliza un tiempo de incubación de 1 h a 30 °C para permitir que se alcance el equilibrio completo. Posteriormente, el radioligando libre se separa del radioligando unido y se cuenta como se describió anteriormente.
|
|
| In vivo |
La absorción de Ticagrelor es rápida con un tmax de 1,3-2 h. Y la Cmax y el área bajo la curva de concentración plasmática-tiempo desde el tiempo 0 hasta el infinito aumentan de manera aparentemente proporcional a la dosis en el rango de dosis estudiado, lo que indica una farmacocinética lineal. La semivida media de la fase terminal (t1/2) es de aproximadamente 7-8,5 h para este compuesto. La inhibición de la agregación plaquetaria (IAP) está relacionada con la dosis y es casi completa a las 2 h con dosis de 100-400 mg. Este químico es bien tolerado, sin eventos adversos graves o relacionados con la dosis ni cambios notables en los valores de laboratorio observados. |
Referencias |
|
Aplicaciones
| Métodos | Biomarcadores | Imágenes | PMID |
|---|---|---|---|
| Western blot | VASP-P / VASP |
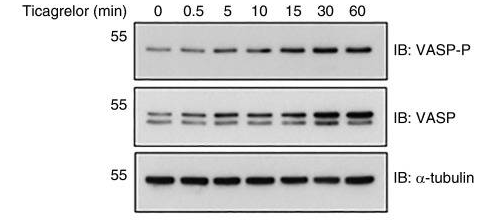
|
27694321 |
Información del ensayo clínico
(datos de https://clinicaltrials.gov, actualizado el 2024-05-22)
| Número NCT | Reclutamiento | Condiciones | Patrocinador/Colaboradores | Fecha de inicio | Fases |
|---|---|---|---|---|---|
| NCT05774431 | Recruiting | Acute Myocardial Infarction |
University Hospital Heidelberg|AstraZeneca |
March 13 2023 | -- |
| NCT05283356 | Recruiting | Severe Aortic Valve Stenosis|Aortic Valve Stenosis|Transcatheter Aortic Valve Replacement (TAVR)|Transcatheter Aortic Valve Implantation (TAVI) |
Fundacin Biomedica Galicia Sur |
January 21 2022 | Phase 4 |
Soporte técnico
Tel: +1-832-582-8158 Ext:3
Si tiene alguna otra consulta, por favor deje un mensaje.
Los productos son solo para uso de investigación. No para uso humano. No vendemos a pacientes.
©Copyright 2013 Selleck Chemicals. Todos los derechos reservados.