solo para uso en investigación
Tariquidar (XR9576) P-gp inhibidor
Cat. No.S8028
Estructura química
Peso molecular: 646.73
Control de calidad
| Dianas relacionadas | CFTR CRM1 CD markers AChR Calcium Channel Sodium Channel Potassium Channel GABA Receptor TRP Channel ATPase |
|---|---|
| Otros P-gp Inhibidores | Zosuquidar 3HCl Elacridar (GF120918) (20S)-Protopanaxadiol Solamargine Sinapine Levistilide A Evodine Valspodar Wilforine Encequidar (HM30181) |
Cultivo celular, tratamiento y concentración de trabajo
| Líneas celulares | Tipo de ensayo | Concentración | Tiempo de incubación | Formulación | Descripción de la actividad | PMID |
|---|---|---|---|---|---|---|
| K562/DOX | Function assay | 1 uM | 10 mins | Inhibition of P-gp in human K562/DOX cells assessed as increase in rhodamine-123 efflux in human K562 cells at 1 uM incubated for 10 mins hrs by flow cytometry relative to untreated control | 28113128 | |
| NB1643 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB1643 cells | 29435139 | |||
| SK-N-SH | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-SH cells | 29435139 | |||
| KB-V1 | Function assay | 200 nM | Inhibition of P-gp in human KB-V1 cells assessed as increase in rhodamine 123 accumulation at 200 nM | 21657271 | ||
| SK-N-MC | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-MC cells | 29435139 | |||
| HepG2 | Cytotoxicity assay | 48 hrs | Cytotoxicity against adriamycin-resistant human HepG2 cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=37.2μM | 27328029 | ||
| K562 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562 cells after 48 hrs by MTT assay, IC50=31.56μM | 28645831 | ||
| K562 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562 cells assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=31.56μM | 29631786 | ||
| A2780adr | Function assay | 10 uM | 30 mins | Inhibition of ABCB1 in human A2780adr cells assessed as increase in accumulation of calcein AM at 10 uM preincubated for 30 mins followed by calcein AM addition measured every 60 secs for 60 mins by fluorescence assay relative to control | 29547272 | |
| K562/A02 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562/A02 cells overexpressing P-gp assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=27.19μM | 29631786 | ||
| CCD-18Co | Cytotoxicity assay | 48 hrs | Cytotoxicity against human CCD-18Co cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| SW620/AD300 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human SW620/AD300 cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| HLF | Cytotoxicity assay | 48 hrs | Cytotoxicity against HLF cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=16.69μM | 27328029 | ||
| CEM/VLB500 | Growth inhibition assay | 3 days | Growth inhibition of human CEM/VLB500 cells after 3 days by resazurin assay, GI50=13.5μM | 17399990 | ||
| MCF7/ADR | Cytotoxicity assay | 48 hrs | Intrinsic cytotoxicity against human MCF7/ADR cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=13.1μM | 27328029 | ||
| HCT116 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human HCT116 cells assessed as cell viability after 48 hrs by MTT assay, IC50=12.5μM | 26197160 | ||
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 12 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=8.28μM | 28645831 | ||
| KBV | Function assay | 5 uM | 72 hrs | Reversal of P-gp-mediated drug resistance in human KBV cells assessed as potentiation of cytotoxicity by measuring IC50 at 5 uM after 72 hrs by MTT assay (Rvb = 398.34 +/- 0.58 uM), IC50=5.24μM | 30384042 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 6 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=4.97μM | 28645831 | ||
| KBV | Function assay | 10 uM | 72 hrs | Reversal of P-gp-mediated drug resistance in human KBV cells assessed as potentiation of cytotoxicity by measuring IC50 at 10 uM after 72 hrs by MTT assay (Rvb = 398.34 +/- 0.58 uM), IC50=4.46μM | 30384042 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured immediately by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=3.02μM | 28645831 | ||
| K562/A02 | Function assay | 5 uM | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 at 5 uM measured after 48 hrs by MTT assay (Rvb = 43.75 to 96.91 uM), IC50=1.97μM | 28645831 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 measured after 48 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=1.6μM | 28645831 | ||
| MCF-7 MX | Function assay | Inhibition of BCRP expressed in MCF-7 MX cells using Hoechst 33342 staining, IC50=1.5μM | 21354800 | |||
| MCF7 | Function assay | Inhibition of ABCG2 in human mitoxantrone-resistant MCF7 cells by Hoechst 33342 assay, IC50=1.44544μM | 18678495 | |||
| HFE | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HFE cells assessed as cell viability after 72 hrs by MTT assay, IC50=1.28μM | 26197160 | ||
| MDCK | Function assay | Inhibition of BCRP expressed in MDCK cells using Hoechst 33342 staining, IC50=0.94μM | 21354800 | |||
| MCF7/Topo | Function assay | Inhibition of ABCG2 overexpressed in human MCF7/Topo cells by flow cytometric-based mitoxantrone efflux assay, IC50=0.916μM | 19170519 | |||
| MCF7/Topo | Function assay | 2 hrs | Inhibition of ABCG2 in human MCF7/Topo cells after 2 hrs by Hoechst 33342 staining based fluorescence assay, IC50=0.526μM | 30128080 | ||
| MCF7/Topo | Function assay | 2 hrs | Inhibition of ABCG2 in human MCF7/Topo cells after 2 hrs by Hoechst 33342 microplate assay, IC50=0.526μM | 24900683 | ||
| MCF7/Topo | Function assay | Inhibition of ABCG2 expressed in human MCF7/Topo cells by Hoechst microplate assay, IC50=0.526μM | 21570282 | |||
| MCF7/Topo | Function assay | Inhibition of ABCG2 in human MCF7/Topo cells by Hoechst 33342 assay, IC50=0.52μM | 26774038 | |||
| KBV1 | Function assay | 10 mins | Inhibition of ABCB1 in human KBV1 cells after 10 mins by Calcein-AM microplate assay, IC50=0.223μM | 24900683 | ||
| Kb-V1 | Function assay | 10 mins | Inhibition of ABCB1 expressed in Kb-V1 cells after 10 mins by calcein-AM assay, IC50=0.223μM | 21570282 | ||
| KBv1 | Function assay | Inhibition of ABCB1 overexpressed in human KBv1 cells by flow cytometric-based calcein-AM efflux assay, IC50=0.223μM | 19170519 | |||
| KBV1 | Function assay | Inhibition of ABCB1 in human KBV1 cells assessed as inhibition of calcein-AM efflux, IC50=0.22μM | 26774038 | |||
| A2780 | Function assay | 30 mins | Inhibition of human Pgp in A2780 cells after 30 mins by Hoechst 33342 assay, IC50=0.12589μM | 18083034 | ||
| KB-3-1 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against human KB-3-1 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.15 +/- 0.04 uM), IC50=0.11μM | 27504669 | |
| OVCAR8 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against human OVCAR8 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.12 +/- 0.03 uM), IC50=0.08μM | 27504669 | |
| A2780/ADR | Function assay | Inhibition of P-glycoprotein-mediated multidrug resistance in adriamycin-resistant human A2780/ADR cells by calcein AM assay, IC50=0.078μM | 19250834 | |||
| A2780adr | Function assay | Inhibition of P-gp expressed in A2780adr cells by calcein AM accumulation assay, IC50=0.08μM | 21354800 | |||
| A2780 | Function assay | Inhibition of P-gp in human adriamycin-resistant A2780 cells by Hoechst 33342 assay, IC50=0.07244μM | 18678495 | |||
| KBV1 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in KBV1 cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 5.07 +/- 0.19 uM), IC50=0.07μM | 27504669 | |
| CEM/VLB500 | Function assay | 3 days | Reversal of P-gp-mediated multidrug resistance to in human CEM/VLB500 cells after 3 days by resazurin assay, EC50=0.068μM | 17399990 | ||
| EMT6/AR1.0 | Function assay | 1 hr | Inhibition of mouse Pgp in EMT6/AR1.0 cells after 1 hr by daunorubicin accumulation assay, IC50=0.06457μM | 18083034 | ||
| EMT6/AR1.0 | Function assay | 1 hr | Inhibition of mouse Pgp in EMT6/AR1.0 cells after 1 hr by daunorubicin accumulation assay, IC50=0.064μM | 18083034 | ||
| MDCK | Function assay | 30 mins | Inhibition of P-glycoprotein (unknown origin) expressed in MDCK cells assessed as reduction of calcein-AM transport after 30 mins by fluorescence assay, EC50=0.044μM | 24607999 | ||
| MDCK | Function assay | 30 mins | Activity at MDR1 (unknown origin) expressed in MDCK cells using calcein AM as substrate incubated for 30 mins prior to substrate addition measured after 30 mins by fluorometric analysis, EC50=0.044μM | 23374872 | ||
| NCI-ADR-RES | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in NCI-ADR-RES cells assessed as potentiation of cytotoxicity by measuring IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 3714.80 +/- 383.58 nM), IC50=0.01851μM | 27504669 | |
| KBV1 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in KBV1 cells assessed as potentiation of cytotoxicity by measuring IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 277.68 +/- 56.61 nM), IC50=0.00066μM | 27504669 | |
| HEK293 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against HEK293 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by CCK8 assay (Rvb = 5.28 +/- 0.74 nM), IC50=0.00495μM | 27504669 | |
| K562/A02 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562/A02 cells after 48 hrs by MTT assay, IC50=27.19μM | 28645831 | ||
| SW620 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human SW620 cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 24 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=14.39μM | 28645831 | ||
| MDCK | Function assay | Inhibition of BCRP expressed in MDCK cells by pheophorbide A assay, IC50=0.85μM | 19932960 | |||
| MCF7 MX | Function assay | Inhibition of BCRP expressed in MCF7 MX cells by Hoechst 33342 staining, IC50=0.68μM | 19932960 | |||
| NCI-ADR-RES | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in NCI-ADR-RES cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 5.54 +/- 0.60 uM), IC50=0.24μM | 27504669 | |
| MDCK | Function assay | Inhibition of MDR1 expressed in MDCK cells using rhodamine 123 staining by flow cytometry, IC50=0.21μM | 21354800 | |||
| CCRF-CEM/VCR1000 | Function assay | 240 secs | Inhibition of P-glycoprotein-mediated daunorubicin efflux from human CCRF-CEM/VCR1000 cells after 240 secs by FACS flow cytometric analysis, IC50=0.03311μM | 22452412 | ||
| HEK293 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 transfected in HEK293 cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by CCK8 assay (Rvb = 504.65 +/- 44.94 nM), IC50=0.02477μM | 27504669 | |
| KB-3-1 | Function assay | 1000 nM | 72 hrs | Potentiation of cytotoxicity against human KB-3-1 cells assessed as IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.78 +/- 0.27 nM), IC50=0.00041μM | 27504669 | |
| OVCAR8 | Function assay | 1000 nM | 72 hrs | Potentiation of cytotoxicity against human OVCAR8 cells assessed as IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 8.53 +/- 1.95 nM), IC50=0.00518μM | 27504669 | |
| MDCK | Function assay | 30 mins | Activity at BCRP (unknown origin) expressed in MDCK cells using rhodamine 123 as substrate incubated for 30 mins prior to substrate addition measured after 30 mins by fluorometric analysis, EC50=0.01μM | 23374872 | ||
| HepG2 | Function assay | 10 uM | 90 mins | Inhibition of P-gp mediated efflux in adriamycin-resistant human HepG2 cells assessed as intracellular rhodamine-123 accumulation at 10 uM incubated in dark condition for 90 mins by flow cytometry relative to control | 27328029 | |
| Haga clic para ver más datos experimentales de líneas celulares | ||||||
Información química, almacenamiento y estabilidad
| Peso molecular | 646.73 | Fórmula | C38H38N4O6 |
Almacenamiento (Desde la fecha de recepción) | |
|---|---|---|---|---|---|
| Nº CAS | 206873-63-4 | Descargar SDF | Almacenamiento de soluciones madre |
|
|
| Sinónimos | XR9576 | Smiles | COC1=C(C=C2CN(CCC2=C1)CCC3=CC=C(C=C3)NC(=O)C4=CC(=C(C=C4NC(=O)C5=CC6=CC=CC=C6N=C5)OC)OC)OC | ||
Solubilidad
|
In vitro |
DMSO
: 8 mg/mL
(12.36 mM)
Water : Insoluble Ethanol : Insoluble |
Calculadora de Molaridad
|
In vivo |
|||||
Calculadora de formulación in vivo (Solución clara)
Paso 1: Introduzca la información a continuación (Recomendado: Un animal adicional para tener en cuenta la pérdida durante el experimento)
Paso 2: Introduzca la formulación in vivo (Esto es solo la calculadora, no la formulación. Por favor, contáctenos primero si no hay una formulación in vivo en la sección de Solubilidad.)
Resultados del cálculo:
Concentración de trabajo: mg/ml;
Método para preparar el líquido maestro de DMSO: mg fármaco predissuelto en μL DMSO ( Concentración del líquido maestro mg/mL, Por favor, contáctenos primero si la concentración excede la solubilidad del DMSO del lote del fármaco. )
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadirμL PEG300, mezclar y clarificar, luego añadirμL Tween 80, mezclar y clarificar, luego añadir μL ddH2O, mezclar y clarificar.
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadir μL Aceite de maíz, mezclar y clarificar.
Nota: 1. Por favor, asegúrese de que el líquido esté claro antes de añadir el siguiente disolvente.
2. Asegúrese de añadir el (los) disolvente(s) en orden. Debe asegurarse de que la solución obtenida, en la adición anterior, sea una solución clara antes de proceder a añadir el siguiente disolvente. Se pueden utilizar métodos físicos como el vórtice, el ultrasonido o el baño de agua caliente para ayudar a la disolución.
Mecanismo de acción
| Targets/IC50/Ki |
P-gp
(CHrB30 cells) 5.1 nM(Kd)
|
|---|---|
| In vitro |
Tariquidar muestra una unión de alta afinidad a la P-gp con una Bmax de 275 pmol/mg. Este compuesto muestra una interacción no competitiva con los sustratos de P-gp. Aumenta la acumulación en estado estacionario de estos citotóxicos en células CHrB30 a niveles observados en células AuxB1 que no expresan P-gp con una EC50 de 487 nM. Este químico es capaz de inhibir la actividad ATPasa sensible al vanadato de la P-gp en un 60-70%, con potentes valores de IC50 de 43 nM. Puede inhibir otros mecanismos de resistencia a concentraciones más altas. 1 µM de este compuesto anula la resistencia mediada por ABCG2 (BCRP) a las camptotecinas in vitro. Potencia la citotoxicidad de varios fármacos, incluida la doxorrubicina; se logra una reversión completa de la resistencia en presencia de 25-80 nM de este químico. En MC26, una línea celular de carcinoma de colon murino con quimiorresistencia intrínseca, la IC50 de doxorrubicina es cinco veces menor en presencia de 0,1 µM de este compuesto (36 frente a 7 nM). En líneas celulares de carcinoma mamario murino, carcinoma de pulmón de células pequeñas humano y carcinoma ovárico humano con resistencia quimioterapéutica adquirida (EMT6/AR1.0, H69/LX4 y 2780 AD), la IC50 de doxorrubicina in vitro es 22-150 veces menor en presencia de 0,1 µM de este químico. La inhibición de P-gp persiste durante 23 h después de su eliminación del sistema de cultivo. Restauró la citotoxicidad de la doxorrubicina en el modelo de esferoide tumoral multicelular NCI/ADRRES (National Cancer Institute) derivado de la línea celular de cáncer de mama MCF7WT. |
| Ensayo de quinasa |
Ensayo de acumulación de fármacos en estado estacionario
|
|
Las células se incuban en un volumen de reacción de 1 mL durante 60 min a 37 ℃ bajo 5% de CO2 para alcanzar el estado estacionario. El efecto de los moduladores XR9576 sobre la acumulación del ligando [3H] se investiga en el rango de concentración de 10-9 - 10-6 M. Este compuesto se añade a partir de una solución madre de DMSO, dando una concentración final de disolvente del 0,2 % (v/v). Después de la recolección celular, el fármaco acumulado se mide mediante recuento por centelleo líquido y se normaliza según el contenido de proteína celular. Las gráficas de la cantidad acumulada en función de la concentración del modulador se ajustan con la ecuación general dosis-respuesta: Y={(a-b)/(1+(X/c)d)}+b, donde: Y=respuesta; a=respuesta inicial; b=respuesta final; c=concentración EC50; d=valor de la pendiente; X=concentración del fármaco.
|
|
| In vivo |
Se ha descubierto que Tariquidar (2-8 mg/kg p.o.) potencia significativamente la actividad antitumoral de la doxorrubicina (5 mg/kg, i.v.) contra el carcinoma de colon murino MC26 in vivo. En xenoinjertos de carcinoma humano, la coadministración de este compuesto (6-12 mg/kg p.o.) restauró completamente la actividad antitumoral contra dos xenoinjertos tumorales humanos MDR altamente resistentes (2780AD, H69/LX4) en ratones nude. |
Referencias |
|
Aplicaciones
| Métodos | Biomarcadores | Imágenes | PMID |
|---|---|---|---|
| Immunofluorescence | MRP7 |
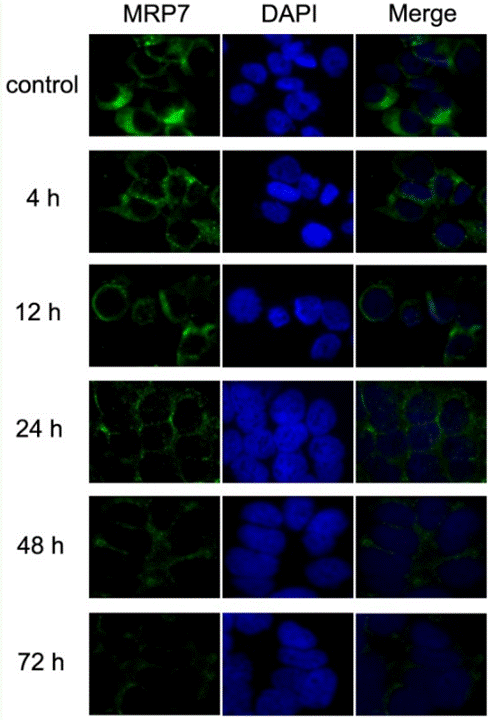
|
23393594 |
Información del ensayo clínico
(datos de https://clinicaltrials.gov, actualizado el 2024-05-22)
| Número NCT | Reclutamiento | Condiciones | Patrocinador/Colaboradores | Fecha de inicio | Fases |
|---|---|---|---|---|---|
| NCT01663545 | Completed | Epilepsies Partial |
National Institute of Neurological Disorders and Stroke (NINDS)|National Institutes of Health Clinical Center (CC) |
July 31 2012 | -- |
| NCT01547754 | Terminated | HIV-Associated Cognitive Motor Complex |
National Institute of Mental Health (NIMH)|National Institutes of Health Clinical Center (CC) |
January 9 2012 | -- |
| NCT01386476 | Completed | Drug Resistance |
National Institute of Mental Health (NIMH)|National Institutes of Health Clinical Center (CC) |
June 15 2011 | -- |
| NCT00082368 | Completed | Cancer |
National Cancer Institute (NCI)|National Institutes of Health Clinical Center (CC) |
May 16 2004 | Phase 2 |
Soporte técnico
Tel: +1-832-582-8158 Ext:3
Si tiene alguna otra consulta, por favor deje un mensaje.
Preguntas frecuentes
Pregunta 1:
Can you please give me more specific and detailed information of how to dissolve and use it (S8028) for in vivo studies?
Respuesta:
It in 30% Propylene glycol, 5% Tween 80, 65% D5W at 30mg/ml will be a suspension or emulsion. If you are going to administrate this compound by oral gavage, it is fine. We also have test some vehicles for it for i.p injection, and it is soluble in 5% DMSO+45% PEG 300+ddH2O at 2mg/ml clearly. When preparing the solution, please dissolve it in DMSO clearly first, then add PEG. After they mixed well, then dilute with water.
Los productos son solo para uso de investigación. No para uso humano. No vendemos a pacientes.
©Copyright 2013 Selleck Chemicals. Todos los derechos reservados.