solo para uso en investigación
Pomalidomide (CC-4047) Agente inmunomodulador
Cat. No.S1567
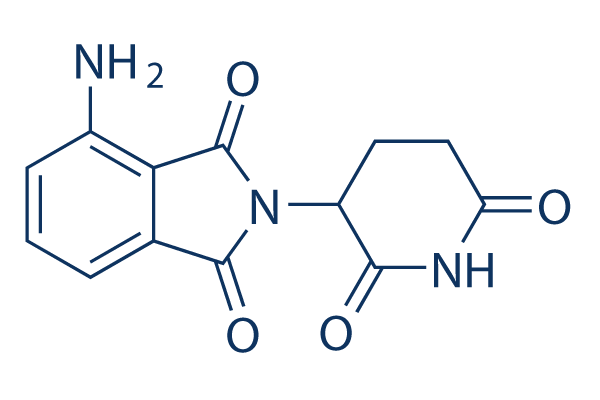
Estructura química
Peso molecular: 273.24
Control de calidad
| Dianas relacionadas | Proteasome E1 Activating E3 Ligase DUB p97 SUMO E2 conjugating |
|---|---|
| Otros E3 ligase Ligand Inhibidores | CC-99282 |
Cultivo celular, tratamiento y concentración de trabajo
| Líneas celulares | Tipo de ensayo | Concentración | Tiempo de incubación | Formulación | Descripción de la actividad | PMID |
|---|---|---|---|---|---|---|
| MOLP-8 | Cytotoxicity Assay | 10 μM | 24 h | potently augments direct and indirect MM cell killing by SAR | 26338273 | |
| J-CD38 | Cytotoxicity Assay | 10 μM | 24 h | potently augments direct and indirect MM cell killing by SAR | 26338273 | |
| R-CD38 | Cytotoxicity Assay | 10 μM | 24 h | potently augments direct and indirect MM cell killing by SAR | 26338273 | |
| BC-3 | Growth Inhibition Assay | 39-1250 nM | 5 d | DMSO | IC50=107 nM, inhibits cell IC50=107 nM, viability dose dependently | 26119939 |
| BCBL-1 | Growth Inhibition Assay | 39-1250 nM | 5 d | DMSO | IC50=74 nM, inhibits cell viability dose dependently | 26119939 |
| JSC-1 | Growth Inhibition Assay | 39-1250 nM | 5 d | DMSO | IC50=34 nM, inhibits cell viability dose dependently | 26119939 |
| VG-1 | Growth Inhibition Assay | 39-1250 nM | 5 d | DMSO | IC50=101 nM, inhibits cell viability dose dependently | 26119939 |
| UMPEL-1 | Growth Inhibition Assay | 39-1250 nM | 5 d | DMSO | IC50=32 nM, inhibits cell viability dose dependently | 26119939 |
| UMPEL-3 | Growth Inhibition Assay | 39-1250 nM | 5 d | DMSO | IC50=111 nM, inhibits cell viability dose dependently | 26119939 |
| BC-1 | Growth Inhibition Assay | 39-1250 nM | 5 d | DMSO | IC50=744 nM, inhibits cell viability dose dependently | 26119939 |
| BCP-1 | Growth Inhibition Assay | 39-1250 nM | 5 d | DMSO | IC50=396 nM, inhibits cell viability dose dependently | 26119939 |
| APK-1 | Growth Inhibition Assay | 39-1250 nM | 5 d | DMSO | IC50=226 nM, inhibits cell viability dose dependently | 26119939 |
| RPMI8226 | Growth Inhibition Assay | 0.01-50 μM | 48 h | DMSO | IC50=8 μM | 26097872 |
| OPM2 | Growth Inhibition Assay | 0.01-50 μM | 48 h | DMSO | IC50=10 μM | 26097872 |
| RPMI8226 | Function Assay | 10 μM | 48 h | DMSO | strengthens cytoplasmic-nuclear shuttling of mTOR and p-mTOR protein | 26097872 |
| OPM2 | Function Assay | 10 μM | 48 h | DMSO | strengthens cytoplasmic-nuclear shuttling of mTOR and p-mTOR protein | 26097872 |
| RPMI8226 | Function Assay | 0.1-10 μM | 4 h | DMSO | increases VEGF mRNA expression | 25053990 |
| SH-SY5Y | Apoptosis Assay | 25 μg/mL | 1 h | causes statistically significant reduction in both CPF- and CPF+CM-induced apoptosis | 24975276 | |
| JJN3 | Growth Inhibition Assay | 0.1-100 μM | 72 h | DMSO | inhibits cell growth slightly | 23178378 |
| XG-1 | Growth Inhibition Assay | 0.1-100 μM | 72 h | DMSO | inhibits cell growth | 23178378 |
| CD138+ | Growth Inhibition Assay | 0.1-100 μM | 72 h | DMSO | inhibits cell growth | 23178378 |
| XG-1 | Function Assay | 2/100 μM | 24 h | DMSO | inhibits CCL3/MIP-1α mRNA expression | 23178378 |
| U266 | Growth Inhibition Assay | 0.01-10 μM | 48 h | DMSO | inhibits cell growth dose dependently | 22552008 |
| CRBN60 | Growth Inhibition Assay | 0.01-10 μM | 48 h | DMSO | inhibits cell growth dose dependently | 22552008 |
| CRNB75 | Growth Inhibition Assay | 0.01-10 μM | 48 h | DMSO | inhibits cell growth dose dependently | 22552008 |
| MM.1S | Growth Inhibition Assay | 0.01-10 μM | 48 h | DMSO | significantly inhibits proliferation at concentrations as low as 0.01μM | 21389327 |
| OPM2 | Growth Inhibition Assay | 0.01-10 μM | 48 h | DMSO | significantly inhibits proliferation at concentrations as low as 0.01μM | 21389327 |
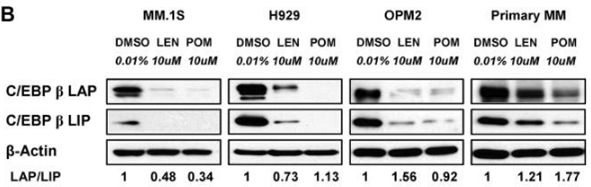
| MM.1S | Function Assay | 10 μM | 72 h | DMSO | significantly decreases the protein level of C/EBPβ isoforms | 21389327 |
| H929 | Function Assay | 10 μM | 72 h | DMSO | significantly decreases the protein level of C/EBPβ isoforms | 21389327 |
| OPM2 | Function Assay | 10 μM | 72 h | DMSO | significantly decreases the protein level of C/EBPβ isoforms | 21389327 |
| CT26 | Function Assay | 1/10 μM | 24 h | reduces the numbers of live colonies | 19638977 | |
| T-cells | Function assay | 2 to 3 days | Inhibition of IL-2 production in human T cells measured after 2 to 3 days by ELISA, EC50 = 0.008 μM. | 23168019 | ||
| DF15 | Function assay | 4 hrs | Induction of cereblon-mediated aiolos degradation in human DF15 cells expressing ePL-tagged aiolos after 4 hrs by luminometric analysis, EC50 = 0.022 μM. | 28425720 | ||
| DF15 | Function assay | 4 hrs | Induction of cereblon-mediated ikaros degradation in human DF15 cells expressing ePL-tagged ikaros after 4 hrs by luminometric analysis, EC50 = 0.024 μM. | 28425720 | ||
| DF15 | Function assay | 4 hrs | Induction of CRL4/CRBN ubiquitin ligase-mediated aiolos degradation in human DF15 cells expressing pLOC-ePL-tagged aiolos after 4 hrs by luminescence based beta-galactosidase enzyme fragmentation complementation assay, EC50 = 0.027 μM. | 28358507 | ||
| NAMALWA | Antiproliferative assay | 72 hrs | Antiproliferative activity against human NAMALWA cells assessed as inhibition of [3H]thymidine incorporation after 72 hrs by scintillation counting, IC50 = 0.03 μM. | 23168019 | ||
| HeLa | Function assay | Inhibition of IL-1-alpha-induced NF-kappaB activation in HeLa cells assessed as blocking of p50/p65 nuclear translocation, IC50 = 1.27 μM. | 17845850 | |||
| DF15 | Function assay | 0.01 to 1 uM | 5 hrs | Induction of cereblon-mediated aiolos degradation in human DF15 cells at 0.01 to 1 uM after 5 hrs by immunoblot analysis | 28425720 | |
| OPM2 | Function assay | 0.01 to 1 uM | 5 hrs | Induction of cereblon-mediated ikaros degradation in human OPM2 cells at 0.01 to 1 uM after 5 hrs by immunoblot analysis | 28425720 | |
| DF15 | Function assay | 0.01 to 1 uM | 5 hrs | Induction of cereblon-mediated ikaros degradation in human DF15 cells at 0.01 to 1 uM after 5 hrs by immunoblot analysis | 28425720 | |
| OPM2 | Function assay | 0.01 to 1 uM | 5 hrs | Induction of cereblon-mediated aiolos degradation in human OPM2 cells at 0.01 to 1 uM after 5 hrs by immunoblot analysis | 28425720 | |
| Haga clic para ver más datos experimentales de líneas celulares | ||||||
Información química, almacenamiento y estabilidad
| Peso molecular | 273.24 | Fórmula | C13H11N3O4 |
Almacenamiento (Desde la fecha de recepción) | |
|---|---|---|---|---|---|
| Nº CAS | 19171-19-8 | Descargar SDF | Almacenamiento de soluciones madre |
|
|
| Sinónimos | CC-4047 | Smiles | C1CC(=O)NC(=O)C1N2C(=O)C3=C(C2=O)C(=CC=C3)N | ||
Solubilidad
|
In vitro |
DMSO
: 100 mg/mL
(365.97 mM)
Water : Insoluble Ethanol : Insoluble |
Calculadora de Molaridad
|
In vivo |
|||||
Calculadora de formulación in vivo (Solución clara)
Paso 1: Introduzca la información a continuación (Recomendado: Un animal adicional para tener en cuenta la pérdida durante el experimento)
Paso 2: Introduzca la formulación in vivo (Esto es solo la calculadora, no la formulación. Por favor, contáctenos primero si no hay una formulación in vivo en la sección de Solubilidad.)
Resultados del cálculo:
Concentración de trabajo: mg/ml;
Método para preparar el líquido maestro de DMSO: mg fármaco predissuelto en μL DMSO ( Concentración del líquido maestro mg/mL, Por favor, contáctenos primero si la concentración excede la solubilidad del DMSO del lote del fármaco. )
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadirμL PEG300, mezclar y clarificar, luego añadirμL Tween 80, mezclar y clarificar, luego añadir μL ddH2O, mezclar y clarificar.
Método para preparar la formulación in vivo: Tomar μL DMSO líquido maestro, luego añadir μL Aceite de maíz, mezclar y clarificar.
Nota: 1. Por favor, asegúrese de que el líquido esté claro antes de añadir el siguiente disolvente.
2. Asegúrese de añadir el (los) disolvente(s) en orden. Debe asegurarse de que la solución obtenida, en la adición anterior, sea una solución clara antes de proceder a añadir el siguiente disolvente. Se pueden utilizar métodos físicos como el vórtice, el ultrasonido o el baño de agua caliente para ayudar a la disolución.
Mecanismo de acción
| Características |
A derivative of thalidomide and up to 10,000 times more potent than thalidomide.
|
|---|---|
| Targets/IC50/Ki |
CRBN
TNF-α
(PBMCs) 13 nM
|
| In vitro |
Pomalidomide inhibe la liberación de TNF-alpha estimulada por lipopolisacáridos (LPS) en PBMC humanos y en sangre total humana con valores de IC50 de 13 nM y 25 nM, respectivamente. Este compuesto inhibe el crecimiento de las células T reguladoras que es estimulado por IL-2 con una IC50 de ~1 μM. El tratamiento con este químico (6,4 nM-10 μM) aumenta la producción de IL-2 en células T de sangre periférica humana, y es ligeramente más potente en el subconjunto CD4+ que en el subconjunto CD8+. Es significativamente más potente que CC-5013 en la elevación de los niveles de IL-2, IL-5 e IL-10, pero solo ligeramente más potente que CC-5013 en la elevación de los niveles de IFN-γ. Este agente mejora la actividad transcripcional de AP-1 inducida por las células SEE y Raji en las células Jurkat de manera dosis-dependiente, con una mejora máxima de 4 veces a 1 μM. La exposición de las células Raji a diversas concentraciones de este compuesto (2,5-40 μg/mL) durante 48 horas conduce a una disminución significativa en la proliferación celular y la síntesis de ADN. Hay una reducción de aproximadamente el 40% en comparación con los controles tratados con vehículo. |
| Ensayo de quinasa |
Inhibición de la síntesis de TNF-α
|
|
La actividad inhibidora de TNF-α se mide en PBMC estimuladas con lipopolisacárido (LPS). Pomalidomide se añade a los PBMC humanos 1 hora antes de la adición de LPS (1 μg/mL) y la incubación se continúa durante 18-20 horas adicionales. Posteriormente se recogen los sobrenadantes y la concentración de TNF-α en los sobrenadantes se determina mediante ELISA. La concentración de este compuesto que inhibe la producción de TNF en un 50 % (IC50) se calcula mediante análisis de regresión no lineal. El ensayo de inhibición de TNF en sangre total humana se realiza de manera similar al ensayo de PBMC, excepto que la sangre total humana fresca heparinada se siembra directamente en placas de microtitulación.
|
|
| In vivo |
Pomalidomide mejora el efecto antitumoral de rituximab contra linfomas de células B en ratones inmunodeficientes combinados graves. La administración de este compuesto en combinación con rituximab, otorga a los ratones un período de supervivencia mediano de 74 días en comparación con los 58 días del tratamiento con CC5013/rituximab y los 45 días de la monoterapia con rituximab. El efecto sinérgico de este compuesto y rituximab puede ser completamente abrogado por la depleción de células NK, apoyando la propuesta de que la expansión de células NK es un mecanismo por el cual este compuesto puede aumentar la actividad antitumoral de rituximab. |
Referencias |
|
Aplicaciones
| Métodos | Biomarcadores | Imágenes | PMID |
|---|---|---|---|
| Western blot | CEBPβ IKZF1 / IKZF3 / UBE2G1 / CRBN |
|
21389327 |
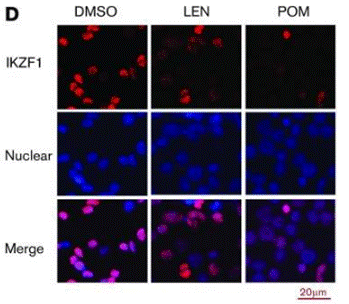
| Immunofluorescence | IKZF1 |
|
29496670 |
Información del ensayo clínico
(datos de https://clinicaltrials.gov, actualizado el 2024-05-22)
| Número NCT | Reclutamiento | Condiciones | Patrocinador/Colaboradores | Fecha de inicio | Fases |
|---|---|---|---|---|---|
| NCT04902443 | Recruiting | Viral Associated Malignancies|Kaposi Sarcoma|EBV/KSHV-associated Lymphomas |
National Cancer Institute (NCI)|National Institutes of Health Clinical Center (CC) |
December 10 2021 | Phase 1 |
Soporte técnico
Tel: +1-832-582-8158 Ext:3
Si tiene alguna otra consulta, por favor deje un mensaje.
Preguntas frecuentes
Pregunta 1:
Is the formulation S1567 in 1% DMSO+30% polyethylene glycol+1% Tween 80 suitable for its oral administration?
Respuesta:
Its suspension in 1% DMSO+30% polyethylene glycol+1% Tween 80 is for oral gavage.
Pregunta 2:
I would like to know if it is racemic or optically active?
Respuesta:
Our S1567 is racemic.
Los productos son solo para uso de investigación. No para uso humano. No vendemos a pacientes.
©Copyright 2013 Selleck Chemicals. Todos los derechos reservados.