Datos técnicos
| Fórmula | C26H31Cl2N7O3 |
||||||
| Peso molecular | 560.48 | Número CAS | 872511-34-7 | ||||
| Solubilidad (25°C)* | In vitro | DMSO | 11 mg/mL (19.62 mM) | ||||
| Water | Insoluble | ||||||
| Ethanol | Insoluble | ||||||
| In vivo (Agregue los solventes al producto individualmente y en orden.) |
|
||||||
|
* <1 mg/ml significa ligeramente soluble o insoluble. * Tenga en cuenta que Selleck prueba la solubilidad de todos los compuestos internamente, y la solubilidad real puede diferir ligeramente de los valores publicados. Esto es normal y se debe a ligeras variaciones entre lotes. * Envío a temperatura ambiente (Las pruebas de estabilidad demuestran que este producto se puede enviar sin medidas de refrigeración.) |
|||||||
Preparación de soluciones madre
Actividad biológica
| Descripción | Infigratinib (BGJ398) es un inhibidor potente y selectivo de FGFR para FGFR1/2/3 con una IC50 de 0,9 nM/1,4 nM/1 nM en ensayos sin células, >40 veces más selectivo para FGFR frente a FGFR4 y VEGFR2, y con poca actividad para Abl, Fyn, Kit, Lck, Lyn y Yes. Fase 2. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Objetivos |
|
||||||||||
| In vitro | Infigratinib (BGJ398) también previene VEGFR2 con baja potencia, con una IC50 de 0,18 μM para la inhibición de este objetivo. Suprime otras quinasas incluyendo ABL, FYN, KIT, LCK, LYN y YES con valores de IC50 de 2,3 μM, 1,9 μM, 0,75 μM, 2,5 μM, 0,3 μM y 1,1 μM, respectivamente. A nivel celular, este compuesto inhibe la proliferación de las células BaF3 dependientes de FGFR1, FGFR2-Q y FGFR3 con IC50 de 2,9 μM, 2,0 μM y 2 μM, respectivamente. Interfiere con la autofosforilación en residuos de tirosina específicos incluyendo FGFR-WT, FGFR2-WT, FGFR3-K650E, FGFR3-S249C y FGFR4-WT con IC50 de 4,6 nM, 4,9 nM, 5 nM, 5 nM y 168 nM, respectivamente. Además, suprime la proliferación de las células cancerosas con sobreexpresión de FGFR3 de tipo salvaje (WT) como RT112, RT4, SW780 y JMSU1 con IC50 de 5 nM, 30 nM, 32 nM y 15 nM, respectivamente. | ||||||||||
| In vivo | En este modelo ortotópico de xenoinjerto de cáncer de vejiga, BGJ398 induce la inhibición del crecimiento tumoral y la estasis después de la administración oral durante 12 días consecutivos a dosis de 10 y 30 mg/kg, respectivamente. Curiosamente, los animales que recibieron este compuesto no mostraron pérdida de peso corporal (10 mg/kg) o un aumento del 10% del peso corporal (30 mg/kg), una indicación adicional de eficacia. Ratas Rowett hembras portadoras de tumores RT112 reciben una única administración oral de la sal monofosfato de Infigratinib (BGJ398) a dosis de 4,25 y 8,51 mg/kg. Disminuye significativamente los niveles de pFRS2 y pMAPK de manera dosis-dependiente. Además, inhibe significativamente la Angiogenesis estimulada por bFGF de manera dosis-dependiente. Sin embargo, no afecta la formación de vasos sanguíneos inducida por VEGF. |
Protocolo (de referencia)
| Ensayo de quinasa: [1] |
|
|---|---|
| Ensayo celular:[1] |
|
| Estudio en animales:[1] |
|
Referencias
|
Validación de productos por parte del cliente

-
Datos de [ Hepatology , 2014 , 59(4), 1427-34 ]
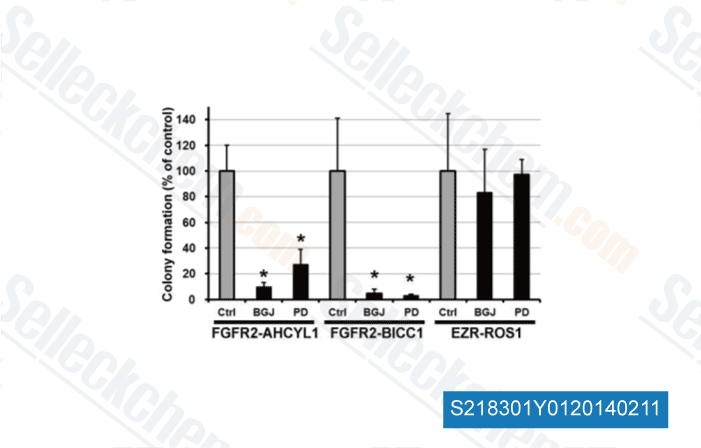
-
Datos de [ Hepatology , 2014 , 59(4), 1427-34 ]

-
Datos de [ J Cell Physiol , 2014 , 229(11), 1647-59 ]

-
, , Oncogene, 2017, 36(27):3831-3841
Sellecks BGJ398 (Infigratinib) Ha sido citado por 256 Publicaciones
| 3D-Printed Scaffolds Promote Enhanced Spinal Organoid Formation for Use in Spinal Cord Injury [ Adv Healthc Mater, 2025, e04817.] | PubMed: 40702833 |
| Selective degradation of FGFR1/2 overcomes antiestrogen resistance in ER+ breast cancer with FGFR1/2 alterations [ Cancer Lett, 2025, 619:217668] | PubMed: 40127812 |
| Abrogation of FGFR signaling blocks β-catenin-induced adrenocortical hyperplasia and aldosterone production [ JCI Insight, 2025, 10(21)e184863] | PubMed: 40956622 |
| Fibroblast growth factor receptor signaling modulates cholesterol storage in a SOAT1-dependent manner to promote mammary tumor cell invasion [ Breast Cancer Res, 2025, 27(1):132] | PubMed: 40665359 |
| Exploiting collateral sensitivity in the evolution of resistance to tyrosine kinase inhibitors in soft tissue sarcomas [ Commun Biol, 2025, 8(1):1185] | PubMed: 40781553 |
| Sonic hedgehog and fibroblast growth factor 8 regulate the evolution of amniote facial proportions [ Commun Biol, 2025, 8(1):84] | PubMed: 39827295 |
| Aspirin-triggered DHA metabolites inhibit angiogenesis [ Front Pharmacol, 2025, 16:1524980] | PubMed: 40070577 |
| Generation of an induced pluripotent stem cell line (NCHi023-A) from a 41-year-old male with nemaline myopathy carrying autosomal dominant ACTA1 c.809-10C>A mutation [ Stem Cell Res, 2025, 85:103701] | PubMed: 40147060 |
| Characterization of an induced pluripotent stem cell line NCHi025-A from a 1-year-old female with pulmonary stenosis harboring a heterozygous HAND2 variant [ Stem Cell Res, 2025, 86:103733] | PubMed: 40378586 |
| Decaying and expanding Erk gradients process memory of skeletal size during zebrafish fin regeneration [ bioRxiv, 2025, 2025.01.23.634576] | PubMed: 39896678 |
POLÍTICA DE DEVOLUCIÓN
La Política de Devolución Incondicional de Selleck Chemical garantiza una experiencia de compra en línea fluida para nuestros clientes. Si no está satisfecho con su compra de alguna manera, puede devolver cualquier artículo(s) dentro de los 7 días posteriores a su recepción. En caso de problemas de calidad del producto, ya sean problemas relacionados con el protocolo o con el producto, puede devolver cualquier artículo(s) dentro de los 365 días a partir de la fecha de compra original. Siga las instrucciones a continuación al devolver productos.
ENVÍO Y ALMACENAMIENTO
Los productos Selleck se transportan a temperatura ambiente. Si recibe el producto a temperatura ambiente, tenga la seguridad de que el Departamento de Inspección de Calidad de Selleck ha realizado experimentos para verificar que la colocación a temperatura normal durante un mes no afectará la actividad biológica de los productos en polvo. Después de la recogida, guarde el producto de acuerdo con los requisitos descritos en la hoja de datos. La mayoría de los productos Selleck son estables en las condiciones recomendadas.
NO PARA USO HUMANO, DIAGNÓSTICO VETERINARIO O TERAPÉUTICO.